Quantitative Cancer Modeling Research Group |
| Staff | Research Interests |
| Philip Hahnfeldt, PhD Principal Investigator |
• Cancer growth dynamics • Radiotherapeutic and chemotherapeutic dosing • DNA damage and repair • Tumor angiogenesis modeling • Gene interaction networks |
| Lynn Hlatky, PhD CCSB Director |
• Cell-cell interactions • Self-organization and emergence • Stability criteria for interaction subpopluations • Evolutionary dynamics |
| Christine E. Briggs, PhD Assistant Investigator |
• Defining microRNA profiles and targets involved in
cancer and response to irradiation • Defining methylation profiles associated with dysregulation of genes/molecular pathways involved in cancer and response to irradiation • Applying aCGH, methylation/SNP-microarray and NGS technologies to identify regions of genomic instability associated with cancer and response to irradiation |
| Sébastien Benzekry, PhD |
• Modeling of metastatic development and tumor-tumor interacations • Modeling of the angiogenesis process • Structured equations in population dynamics • Scheduling optimization for anti-cancerous therapies |
| Xuefeng (Ryan) Gao, PhD Postdoctoral Fellow |
• Modeling tumor growth, tumor angiogenesis, and invasion • Darwin evolution of cancer cells: the emergence and distribution of cancer cell phenotypes • Modeling cancer gene therapies (e.g., virotherapy, bacterial therapy) • Dynamics of multi-cellular systems • Cell-centered multi-scale modeling and simulation • Biomedical visualizations, animations and movies • Interactive technologies |
| Rainer Sachs, PhD Associate Investigator |
• Radiation-induced carcinogenesis • Chronic myeloid leukemia (CML) • Cosmic radiation risk estimation for astronauts • Second cancers after radiotherapy • Radiation-induced chromosome aberrations • Large-scale geometry of chromatin during cell cycle interphase |
| Kathleen Wilkie, PhD Assistant Investigator |
• Cell adhesion and the extracellular matrix • Tissue-matrix remodelling • The effects of mechanical stresses on cells • Heterogeneous cell populations in cancer progression • Growth, development, and aging effects on biological tissues • Mechanical properties of biological tissues |
Mathematical Research at the CCSB
 Quantitative research at the CCSB embraces the principle that cancer is not just a disease of cells, but is facilitated at the population and inter-tissue levels. Accordingly, a multi-level approach must be implemented to fully understand its origin and course. Initial events in carcinogenesis occur within cells at the molecular level as repair and proliferation dysfunctions, leading to genomic instability, aneuploidy, and final transformation. But after cancer cell creation, the clone advances to encounter additional molding and fate-determining events defined by cell-cell interactions. The associated stroma (fibroblasts and extracellular matrix) plays a critical role in cancer progression, as does induced tumor vascularization (angiogenesis). Both act to determine whether a nascent cancer advances to become symptomatic disease. Without stromal activation and angiogenesis, tumor development is halted early. Under NASA Specialized Centers of Research (NSCOR) funding, these studies are focusing on the determination of cancer risk to astronauts who will be exposed to harmful solar particle events (SPEs) and galactic cosmic radiations (GCRs) during long-term space flight. By extension, we hope to better understand the carcinogenesis process more generally, and discover new therapeutic interventions for improved cancer treatment.
Quantitative research at the CCSB embraces the principle that cancer is not just a disease of cells, but is facilitated at the population and inter-tissue levels. Accordingly, a multi-level approach must be implemented to fully understand its origin and course. Initial events in carcinogenesis occur within cells at the molecular level as repair and proliferation dysfunctions, leading to genomic instability, aneuploidy, and final transformation. But after cancer cell creation, the clone advances to encounter additional molding and fate-determining events defined by cell-cell interactions. The associated stroma (fibroblasts and extracellular matrix) plays a critical role in cancer progression, as does induced tumor vascularization (angiogenesis). Both act to determine whether a nascent cancer advances to become symptomatic disease. Without stromal activation and angiogenesis, tumor development is halted early. Under NASA Specialized Centers of Research (NSCOR) funding, these studies are focusing on the determination of cancer risk to astronauts who will be exposed to harmful solar particle events (SPEs) and galactic cosmic radiations (GCRs) during long-term space flight. By extension, we hope to better understand the carcinogenesis process more generally, and discover new therapeutic interventions for improved cancer treatment.
The origin of cancer may be attributed to events that alter the repair machinery of the cell and destabilize its genome. Of active interest in this regard is how nucleotide-level DNA damage and repair translates into chromosome aberrations, as chromosome-level misrepair is the feature most closely identified with carcinogenic transformation. By developing a revised theory for double-strand break repair/misrepair following ionizing radiation, we have found it possible to improve upon the commonly-accepted repair models, thus providing a better template for surrogate radiation cancer risk estimation.
We have gone on to associate radiation action to the first population-level bottleneck limiting the growth of hyperplastic clones through a process of self-limited cell growth. The resulting deterministic model for early carcinogenesis has proven to be competitive with the current stochastic standard in explaining major epidemiological data sets on radiation-induced carcinogenesis.
A second bottleneck encountered early in carcinogenesis is nutrient availability. Without angiogenesis, a tumor cannot grow beyond approximately 1mm in size. This obstacle to tumor growth requires the development of angiogenic potential within the growing clone of tumor cells — an ‘angiogenic switch’. Interestingly, it was discovered quite by accident some time ago while performing autopsies on adults who died of non-cancer causes (Black and Welch, 1993 and Black and Welch, 2010) that most middle-aged people harbor dormant, non-threatening cancer lesions. Held back by the failure to initiate angiogenesis, the tumors remained harmless throughout these individuals' lifetimes. One major objective of our Center is to understand how such a stasis may be maintained, and perhaps re-established, as part of a novel therapeutic approach. We have made inroads into quantitatively understanding this natural control in tumor growth and how antiangiogenic therapy might best be applied to achieve this goal.
 Basic Cell Kinetics, Cancer Stem Cells, and Tumor Morphology
Basic Cell Kinetics, Cancer Stem Cells, and Tumor Morphology
Tumors are intrinsically heterogeneous. The majority of the tumor cells have limited life span and replicative potential, and only a small minority (so-called cancer stem cells) live forever, divide infinitely and potentially produce more such stem cells. It is these stem cells that determine tumor formation, and their dynamics is counterintutively inhibited by their non-stem progeny. Only a high migration rate can liberate stem cells and enable their migration to seed new clones in the vicinity of the original cluster. In this manner, the tumour continually ‘self-metastasizes’.
We use cellular automaton models to define the behavior of single cells, and then let single cells populate a compuational domain. As the number of cancer cell increases over time competition for environmental resources (such as space) defines population dynamics. A result is a cancer cell population (a tumor) growing sub-exponentially. Tumor progession is dictated by the ability of stem cells to form self-metastases that together form a malignant invasive morphology.
Modeling DNA Damage and Repair and Chromosome Geometry

The chromosome damage/repair studies have interrelated results on cytogenetics and interphase chromosome localization and geometry. By developing a revised theory for double-strand break repair/misrepair following ionizing radiation, we found it possible to improve upon the commonly-accepted repair models, in the process obtaining information on how chromosomes are packaged within the nucleus. Incomplete exchange models developed from these studies were also able to explain the relation between acentric and dicentric counts and the excess dispersion (variance/mean) for the number of acentric fragments relative to dicentrics seen in human lymphocytes exposed to various radiation types and doses.
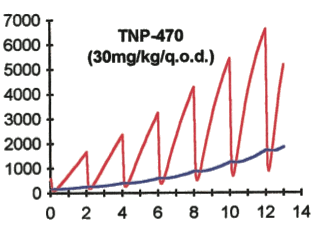 Angiogenesis Modeling and Anti-angigenic Therapy
Angiogenesis Modeling and Anti-angigenic Therapy
Extending from collaboration with the late Dr. Judah Folkman of Children's Hospital Boston, we are exploring the unique tumor/vascular regression kinetics of anti-angiogenic therapy. The indirect means by which tumor suppression is here accomplished, coupled with the recent finding that tumors both stimulate and inhibit their own vascularization, points to a need to formulate tumor-vascular models that properly capture the dynamics. We use sets of differential equations that simultaneously consider vascular response to anti-angiogenic agents and subsequent suppression of tumor growth. The result is a formalism that is proving to be both explanatory and clinically predictive. This work is an example of quantitative translational research, a “workstation-to-bench-to-bedside” research strategy embraced by the CCSB. An important dynamic to surface from these studies is the self-imposed Gompertz restriction on growth imposed by a tumor on itself. Implications for the general organogenic control of tissue mass are suggested. Another study of dose rate effects addresses an as yet un-quantified effect — the utility of so-called “metronomic” (small, evenly-spaced) dosing on the treatment response of a tumor population. It was shown considering the response of a heterogeneous target to various dosing protocols that: 1) metronomic dosing does indeed offer the best tumor suppression, and 2) the shift to metronomic dosing from more traditional “up-front” dosing regimens favors the endothelial cell compartment. The theory offers one explanation for numerous reports of an antiangiogenic response using the metronomic scheme.
Tumor Heterogeneity
 A unifying theme in both the DNA repair and angiogenesis studies is the role tumor heterogeneity and inter-tissue interactions play in carcinogenesis risk and cancer treatment response. Under our NASA NSCOR Program Project dedicated to examining how inter-cellular interactions modulate carcinogenesis, we are currently employing quantitative methods to understand the unifying mechanisms more precisely. A deterministic carcinogenesis risk model that incorporates cell-cell interactions was developed which compares favorably to the current stochastic model standard in explaining epidemiologic data on atom bomb survivors. Additional unpublished studies are now showing that inter-cellular interactions may even decide the course of cancer after the fact of tumor cell creation, proving these interactions to be a vital augment to current cell-centric focuses on cancer origin and treatment response.
A unifying theme in both the DNA repair and angiogenesis studies is the role tumor heterogeneity and inter-tissue interactions play in carcinogenesis risk and cancer treatment response. Under our NASA NSCOR Program Project dedicated to examining how inter-cellular interactions modulate carcinogenesis, we are currently employing quantitative methods to understand the unifying mechanisms more precisely. A deterministic carcinogenesis risk model that incorporates cell-cell interactions was developed which compares favorably to the current stochastic model standard in explaining epidemiologic data on atom bomb survivors. Additional unpublished studies are now showing that inter-cellular interactions may even decide the course of cancer after the fact of tumor cell creation, proving these interactions to be a vital augment to current cell-centric focuses on cancer origin and treatment response.
BioInformatics
Our BioInformatics and Network Statistics Group uses high-throughput data from both in-house and external data sources to investigate molecular targets, molecular pathways, and molecular networks to inform our clinical and biological staff concering potential new targets and new biology. In addition, we are investigating the interaction of -omics data with cellular macroscopic images to go beyond the conventional views of molecular and cellular biology.
Resources
Software:
Chromosome Aberration Simulator (CAS)
Software for simulating radiogenic aberrations in detail. Executables and source code are freely available for any educational or research purpose.
Chromosome Aberration Analyzer (CAA)
Software for quantitative analyses of observed or simulated karyotypes.
Representative Publications:
The following articles on the topic of quantitative modeling have been
published by researchers at CCSB:
- Benzekry S, Beheshti A, Hahnfeldt P, Hlatky L. Capturing the driving role of tumor-host crosstalk in a dynamical model of tumor growth. Bio-Protocol. 2015 Nov 5;5(21):e1644.
- Poleszczuk J, Hahnfeldt P, Enderling H. Therapeutic implications from sensitivity analysis of tumor angiogenesis models. PLoS One. 2015 Mar 18;10(3):e0120007.
- Poleszczuk J, Hahnfeldt P, Enderling H. Evolution and phenotypic selection of cancer stem cells. PLoS Comput Biol. 2015 Mar 5;11(3):e1004025.
- Radivoyevitch T, Siranart N, Hlatky L, Sachs R. Stochastic process pharmacodynamics: dose timing in neonatal gentamicin therapy as an example. AAPS J. 2015 Mar;17(2):447-56. Epub 2015 Feb 7.
- Benzekry S, Lamont C, Beheshti A, Tracz A, Ebos JM, Hlatky L, Hahnfeldt P. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol. 2014 Aug 28;10(8):e1003800. eCollection 2014 Aug.
- Weekes SL, Barker B, Bober S, Cisneros K, Cline J, Thompson A, Hlatky L, Hahnfeldt P, Enderling H. A multicompartment mathematical model of cancer stem cell-driven tumor growth dynamics. Bull Math Biol. 2014 Jul;76(7):1762-82. Epub 2014 May 20.
- Enderling H, Chaplain MJ. Mathematical modeling of tumor growth and treatment. Curr Pharm Des. 2014;20(30):4934-40. Epub 2013 Nov 25.
- Poleszczuk J, Hahnfeldt P, Enderling H. Biphasic modulation of cancer stem cell-driven solid tumour dynamics in response to reactivated replicative senescence. Cell Prolif. 2014 Jun;47(3):267-76. Epub 2014 Mar 25.
- Gao X, McDonald JT, Naidu M, Hahnfeldt P, Hlatky L. A proposed quantitative index for assessing the potential contribution of reprogramming to cancer stem cell kinetics. Stem Cells Int. 2014 May 12;2014(article ID 249309);1-8. Epub 2014 Apr 17.
- Radivoyevitch T, Jankovic GM, Tiu RV, Saunthararajah Y, Jackson RC, Hlatky LR, Gale RP, Sachs RK. Sex differences in the incidence of chronic myeloid leukemia. Radiat Environ Biophys. 2014 Mar;53(1):55-63. Epub 2013 Dec 13.
- Benzekry S, Gandolfi A, Hahnfeldt P. Global dormancy of metastases due to systemic inhibition of angiogenesis. PLoS One. 2014 Jan 21;9(1):e84249. eCollection 2014 Jan 21.
- Brenner D, Shuryak I, Sachs RK. Radiotherapy-Induced Carcinogenesis and Leukemogenesis: Mechanisms and Quantitative Modeling. In: Rubin P, Constine LS, Marks LB (eds). ALERT – Adverse Late Effects of Cancer Treatment. Volume 1: General Concepts and Specific Precepts. New York: Springer, 2014:205-226. In Series: Medical Radiology – Radiation Oncology: Brady LW, Heilmann H-P, Molls M, Nieder C (series eds).
- Fakir H, Hlatky L, Li H, Sachs R. Repopulation of interacting tumor cells during fractionated radiotherapy: Stochastic modeling of the tumor control probability. Med Phys. 2013 Dec;40(12):121716. Epub 2013 Nov 15. EDITOR'S PICK selection
- Benzekry S, Hahnfeldt P. Maximum tolerated dose versus metronomic scheduling in treatment of metastatic cancers. J Theor Biol. 2013 Oct 21;335:235-44. Epub 2013 Jul 10.
- McGuire MF, Enderling H, Wallace DI, Batra J, Jordan M, Kumar S, Panetta JC, Pasquier E. Formalizing an integrative, multidisciplinary cancer therapy discovery workflow. Cancer Res. 2013 Oct 15;73(20):6111-7. Epub 2013 Aug 16.
- Wilkie KP, Hahnfeldt P. Mathematical models of immune-induced cancer dormancy and the emergence of immune evasion. Interface Focus. 2013 Aug 6;3(4):20130010. Epub 2013 Jun 25.
- Wilkie KP, Hahnfeldt P. Tumor–immune dynamics regulated in the microenvironment inform the transient nature of immune-induced tumor dormancy. Cancer Res. 2013 Jun 15;73(12):3534-44. Epub 2013 Mar 27.
- Rietman EA, Friesen DE, Hahnfeldt P, Hlatky L, Tuszynski JA. An integrated multidisciplinary model describing initiation of cancer and the Warburg hypothesis. Theor Biol Med Model. 2013 Jun 10;10:39.
- Wilkie KP, Hahnfeldt P. Modeling the Dichotomy of the Immune Response to Cancer: Cytotoxic Effects and Tumor-Promoting Inflammation. arXiv:1305.3634 [q-bio.CB]. 2013 May 15.
- Hahnfeldt P, Hlatky L, Klement GL. Center of cancer systems biology second annual workshop — tumor metronomics: timing and dose level dynamics. Cancer Res. 2013 May 15;73(10):2949-54. Epub 2013 Mar 14.
- Kareva I, Hahnfeldt P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: distinct or linked? Cancer Res. 2013 May 1;73(9):2737-42. Epub 2013 Feb 19.
- Enderling H, Hlatky L, Hahnfeldt P. Cancer stem cells: a minor cancer subpopulation that redefines global cancer features. Front Oncol. 2013 Apr 15;3:76. Epub 2013 Apr 15.
- Lignet F, Benzekry S, Wilson S, Billy F, Saut O, Tod M, You B, Adda Berkane A, Kassour S, Wei MX, Grenier E, Ribba B. Theoretical investigation of the efficacy of antiangiogenic drugs combined to chemotherapy in xenografted mice. J Theor Biol. 2013 Mar 7;320:86-99. Epub 2012 Dec 21.
- Gao X, McDonald JT, Hlatky L, Enderling H. Acute and fractionated irradiation differentially modulate glioma stem cell division kinetics. Cancer Res. 2013 Mar 1;73(5):1481-90. Epub 2012 Dec 26.
- Kareva I. Biological Stoichiometry in Tumor Micro-environments. PLoS One. 2013;8(1):e51844. Epub 2013 Jan 22.
- Hillen T, Enderling H, Hahnfeldt P. The tumor growth paradox and immune system-mediated selection for cancer stem cells. Bull Math Biol. 2013 Jan;75(1):161-84. Epub 2012 Nov 30.
- Gao X, McDonald JT, Hlatky L, Enderling H. Cell-Cell Interactions in Solid Tumors — the Role of Cancer Stem Cells. In: D'Onofrio A, Cerrai P, Gandolfi A (eds). New Challenges for Cancer Systems Biomedicine. Italy: Springer, 2012: 191-204. In Series: SIMAI Springer Series: Bellomo N, Formaggia L, Bangerth W, Nobile F, Pareschi L, Tercero PP, Tosin A, Zubelli JP (series Eds).
- Hahnfeldt P. The host support niche as a control point for tumor dormancy: implications for tumor development and beyond. Adv Exp Med Biol. 2013;734:19-35. doi: 10.1007/978-1-4614-1445-2_2. (also in Systems Biology of Tumor Dormancy. Enderling H, Almog N, Hlatky L, editors. New York: Springer; 2013: 291p.)
- Enderling H. Cancer stem cells and tumor dormancy. Adv Exp Med Biol. 2013,734:55-71. doi: 10.1007/978-1-4614-1445-2_4. (also in Systems Biology of Tumor Dormancy. Enderling H, Almog N, Hlatky L, editors. New York: Springer, 2013: 291p.)
- Wilkie KP. A review of mathematical models of cancer-immune interactions in the context of tumor dormancy. Adv Exp Med Biol. 2013;734:201-34. doi: 10.1007/978-1-4614-1445-2_10. (also in Systems Biology of Tumor Dormancy. Enderling H, Almog N, Hlatky L, editors. New York: Springer, 2013: 291p.)
- Enderling H, Hlatky L, Hahnfeldt P. Immunoediting: Evidence of the multifaceted role of the immune system in self-metastatic tumor growth. Theor Biol Med Model. 2012 Jul 28;9(1):31. Epub 2012 July 28.
- Breitkreutz D, Hlatky L, Rietman E, Tuszynski JA. Molecular signaling network complexity is correlated with cancer patient survivability. Proc Natl Acad Sci U S A. 2012 Jun 5;109(23):9209-12. Epub 2012 May 21.
- Radivoyevitch T, Hlatky L, Landaw J, Sachs RK. Quantitative modeling of chronic myeloid leukemia: insights from radiobiology. Blood. 2012 May 10;119(19):4363-71. Epub 2012 Feb 21. COVER ARTICLE
- Enderling H, Hahnfeldt P, Hlatky L, Almog N. Systems biology of tumor dormancy: linking biology and mathematics on multiple scales to improve cancer therapy. Cancer Res. 2012 May 1;72(9):2172-5. Epub 2012 Mar 13.
- Wilkie KP, Drapaca CS, Sivaloganathan S. A mathematical investigation of the role of intracranial pressure pulsations and small gradients in the pathogenesis of hydrocephalus. International Journal of Numerical Analysis and Modeling, Series B. 3(1):36-51, 2012.
- Wilkie KP, Nagra G, Johnston M. A mathematical analysis of physiological and molecular mechanisms that modulate pressure gradients and facilitate ventricular expansion in hydrocephalus. International Journal of Numerical Analysis and Modeling, Series B. 3(1):65-81, 2012.
- Benzekry S. Mathematical and numerical analysis of a model for anti-angiogenic therapy in metastatic cancers. Esaim Math Model Numer Anal. 2012 Mar;46(2):207-37. Epub 2011 Oct 6.
- Enderling H, Hlatky L, Hahnfeldt P. The promoting role of a tumour-secreted chemorepellent in self-metastatic tumour progression. Math Med Biol. 2012 Mar;29(1):21-9. Epub 2010 Oct 6.
- Benzekry S. Passing to the limit 2D-1D in a model for metastatic growth. J Biol Dyn. 2012;6 Suppl 1:19-30. Epub 2011 Jun 24.
- Benzekry S, André N, Benabdallah A, Ciccolini J, Faivre C, Hubert F, Barbolosi D. Modeling the impact of anticancer agents on metastatic spreading. Math Model Natl Phenom. 2012 Jan;7(1):306-36. Epub 2012 Jan 25.
- Benzekry S, Chapuisat G, Ciccolini J, Erlinger A, Hubert F. A new mathematical model for optimizing the combination between antiangiogenic and cytotoxic drugs in oncology / Un nouveau modèle mathématique pour l'optimisation des combinaisons antiangiogéniques/cytotoxiques en cancérologie. Comptes Rendus Mathematique. 2012 Jan;350(1-2):23-8. Epub 2011 Dec 16.
- Benzekry S. Mathematical analysis of a two-dimensional population model of metastatic growth including angiogenesis. J Evol Equ 11(1): 187-213, 2011. Epub 2010 Dec 23.
- Morton CI, Hlatky L, Hahnfeldt P, Enderling H. Non-stem cancer cell kinetics modulate solid tumor progression. Theor Biol Med Model. 2011 Dec 30;8:48.
- Rietman EA, Karp RL, Tuszynski JA. Review and application of group theory to molecular systems biology. Theor Biol Med Model. 2011 Jun 22;8:21.
- Rietman EA, Colt JZ, Tuszynski JA. Interactomes, manufacturomes and relational biology: analogies between systems biology and manufacturing systems. Theor Biol Med Model. 2011 Jun 20;8:19.
- Enderling H, Hahnfeldt P. Cancer stem cells in solid tumors: is ‘evading apoptosis’ a hallmark of cancer? Prog Biophys Mol Biol. 2011 Aug;106(2):391-9. Epub 2011 Apr 5.
- Tang J, Enderling H, Becker-Weimann S, Pham C, Polyzos A, Chen CY, Costes SV. Phenotypic transition maps of 3D breast acini obtained by imaging-guided agent-based modeling. Integr Biol (Camb). 2011 Apr;3(4):408-21. Epub 2011 Mar 4. COVER ARTICLE
- Sachs RK, Johnsson K, Hahnfeldt P, Luo J, Chen A, Hlatky L. A multicellular basis for the origination of blast crisis in chronic myeloid leukemia. Cancer Res. 2011 Apr 15;71(8):2838-47. Epub 2011 Apr 12.
- Benzekry S. Mathematical analysis of a two-dimensional population model of metastatic growth including angiogenesis. J Evol Equ. 2011 Mar 1;11(1):187-213. Epub 2010 Dec 23.
- Shuryak I, Sachs RK, Brenner DJ. A new view of radiation-induced cancer. Radiat Prot Dosimetry. 2011 Feb;143(2-4):358-64. Epub 2010 Nov 27.
- Enderling H, Chaplain MA, Hahnfeldt P. Quantitative modeling of tumor dynamics and radiotherapy. Acta Biotheor. 2010 Dec;58(4):341-53. Epub 2010 Jul 24.
- Shuryak I, Sachs RK, Brenner DJ. Cancer risks after radiation exposure in middle age. J Natl Cancer Inst. 2010 Nov 3;102(21):1628-36. Epub 2010 Oct 25.
- Shuryak I, Ullrich RL, Sachs RK, Brenner DJ. The balance between initiation and promotion in radiation-induced murine carcinogenesis. Radiat Res. 2010 Sep;174(3):357-66.
- Enderling H, Hlatky L, Hahnfeldt P. Tumor morphological evolution: directed migration and gain and loss of the self-metastatic phenotype. Biol Direct. 2010 Apr 20;5:23.
- Fakir H, Hofmann W, Sachs RK. Modeling progression in radiation-induced lung adenocarcinomas. Radiat Environ Biophys. 2010 May;49(2):169-176. Epub 2010 Jan 8.
- Enderling H, Anderson AR, Chaplain MA, Beheshti A, Hlatky L, Hahnfeldt P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009 Nov 15;69(22):8814-21. Epub 2009 Nov 3.
- Fakir H, Tan WY, Hlatky L, Hahnfeldt P, Sachs RK. Stochastic population dynamic effects for lung cancer progression. Radiat Res. 2009 Sep;172(3):383-93.
- Shuryak I, Hahnfeldt P, Hlatky L, Sachs RK, Brenner DJ. A new view of radiation-induced cancer: integrating short- and long-term processes. Part I: approach. Radiat Environ Biophys. 2009 Aug;48(3):263-74.
- Shuryak I, Hahnfeldt P, Hlatky L, Sachs RK, Brenner DJ. A new view of radiation-induced cancer: integrating short- and long-term processes. Part II: second cancer risk estimation. Radiat Environ Biophys. 2009 Aug;48(3):275-86.
- Enderling H, Hlatky L, Hahnfeldt P. Migration rules: tumours are conglomerates of self-metastases. Br J Cancer. 2009 Jun 16;100(12):1917-25.
- Enderling H, Park D, Hlatky L, Hahnfeldt P. The importance of spatial distribution of stemness and proliferation state in determining tumor radioresponse. Math Model Nat Phenom. 2009;4(3):117-33.
- Fakir H, Hofmann W, Tan WY, Sachs RK. Triggering-response model for radiation-induced bystander effects. Radiat Res. 2009 Mar;171(3):320-31.
- Brenner D, Shuryak I, Sachs R. Radiotherapy-Induced Carcinogenesis and Leukemogenesis: Mechanisms and Quantitative Modeling. In: ALERT Adverse Late Effects of Cancer Treatment. Volume 1: General Concepts and Principles (P. Rubin, L. S. Constine, L. B. Marks, J. P. Williams, and J. T. Hansen, eds.), Springer, New York. In Series: Medical Radiology -- Radiation Oncology, Brady LW, Heilmann H-P, Molls M, Nieder C (Series Editors), 2009.
- Li L, McCormack AA, Nicholson JM, Fabarius A, Hehlmann R, Sachs RK, Duesberg PH. Cancer-causing karyotypes: chromosomal equilibria between destabilizing aneuploidy and stabilizing selection for oncogenic function. Cancer Genet Cytogenet. 2009 Jan 1;188(1):1-25.
- Enderling H, Alexander NR, Clark ES, Branch KM, Estrada L, Crooke C, Jourquin J, Lobdell N, Zaman MH, Guelcher SA, Anderson AR, Weaver AM. Dependence of invadopodia function on collagen fiber spacing and cross-linking: computational modeling and experimental evidence. Biophys J. 2008 Sep;95(5):2203-18.
- Feinendegen L, Hahnfeldt P, Schadt EE, Stumpf M, Voit EO. Systems biology and its potential role in radiobiology. Radiat Environ Biophys. 2008 Feb;47(1):5-23.
- Piotrowska MJ, Enderling H, an der Heiden U, Mackey MC. Mathematical modeling of stem cells related to cancer. In: Dittmar T, Zänker KS (eds). Cancer and Stem Cells. Nova Science Publishers, Inc., 2008:11-35.
- Shuryak I, Sachs RK, Brenner DJ. Biophysical models of radiation bystander effects: 1. Spatial effects in three-dimensional tissues. Radiat Res. 2007 Dec;168(6):741-9.
- Sachs RK, Shuryak I, Brenner D, Fakir H, Hlatky L, Hahnfeldt P. Second cancers after fractionated radiotherapy: stochastic population dynamics effects. J Theor Biol. 2007 Dec 7;249(3):518-31.
- Hodgson DC, Koh ES, Tran TH, Heydarian M, Tsang R, Pintilie M, Xu T, Huang L, Sachs RK, Brenner DJ. Individualized estimates of second cancer risks after contemporary radiation therapy for Hodgkin lymphoma. Cancer. 2007 Dec 1;110(11):2576-86.
- Brenner DJ, Shuryak I, Russo S, Sachs RK. Reducing second breast cancers: a potential role for prophylactic mammary irradiation. J Clin Oncol. 2007 Nov 1;25(31):4868-72.
- Levy D, Reeder C, Loucas B, Hlatky L, Chen A, Cornforth M, Sachs R. Interpreting chromosome aberration spectra. J Comput Biol. 2007 Mar;14(2):144-55.
- Ponomarev AL, Belli M, Hahnfeldt PJ, Hlatky L, Sachs RK, Cucinotta FA. Subtraction of background damage in PFGE experiments on DNA fragment-size distributions. Radiat Environ Biophys. 2007 Jun;46(2):155-60.
- Koh ES, Tran TH, Heydarian M, Sachs RK, Tsang RW, Brenner DJ, Pintilie M, XuT, Chung J, Paul N, Hodgson DC. A comparison of mantle versus involved-field radiotherapy for Hodgkin's lymphoma: reduction in normal tissue dose and second cancer risk. Radiat Oncol. 2007 Mar 15;2:13.
- Shuryak I, Sachs RK, Hlatky L, Little MP, Hahnfeldt P, Brenner DJ. Radiation-induced leukemia at doses relevant to radiation therapy: modeling mechanisms and estimating risks. J Natl Cancer Inst. 2006 Dec 20;98(24):1794-806.
- Ponomarev AL, Belli M, Hahnfeldt PJ, Hlatky L, Sachs RK, Cucinotta FA. A robust procedure for removing background damage in assays of radiation-induced DNA fragment distributions. Radiat Res. 2006 Dec;166(6):908-16.
- Fakir H, Sachs RK, Stenerlöw B, Hofmann W. Clusters of DNA double-strand breaks induced by different doses of nitrogen ions for various LETs: experimental measurements and theoretical analyses. Radiat Res. 2006 Dec;166(6):917-27.
- Brenner DJ, Sachs RK. Estimating radiation-induced cancer risks at very low doses: rationale for using a linear no-threshold approach. Radiat Environ Biophys. 2006 Mar;44(4):253-6.
- Plan Y, Hlatky L, Hahnfeldt P, Sachs R, Loucas B, Cornforth M. Full-color painting reveals an excess of radiation-induced dicentrics involving homologous chromosomes. Int J Radiat Biol. 2005 Aug;81(8):613-20.
- Li R, Hehlman R, Sachs R, Duesberg P. Chromosomal alterations cause the high rates and wide ranges of drug resistance in cancer cells. Cancer Genet Cytogenet. 2005 Nov;163(1):44-56.
- Sachs RK, Brenner DJ. Solid tumor risks after high doses of ionizing radiation. Proc Natl Acad Sci USA. 2005 Sep 13;102(37):13040-5. Epub 2005 Sep 6.
- Sachs RK, Chan M, Hlatky L, Hahnfeldt P. Modeling intercellular interactions during carcinogenesis. Radiat Res. 2005 Sep;164(3):324-31.
- Sachs RK, Brenner DJ. Chromosome Aberrations Produced by Ionizing Radiation: Quantitative Studies. Beta-test version of part of an NCBI Bookshelf Internet Textbook; PubMed, National Center for Biotechnology Information, NLM, NIH. http://radiobiology.math.berkeley.edu/, 2005.
- Vives S, Loucas B, Vazquez M, Brenner DJ, Sachs RK, Hlatky L, Cornforth M, Arsuaga J. SCHIP: statistics for chromosome interphase positioning based on interchange data. Bioinformatics. 2005 Jul 15;21(14):3181-2.
- Mestres M, Caballin MR, Schmid E, Stephan G, Sachs R, Barrios L, Barquinero JF. Analysis of alpha-particle induced chromosome aberrations in human lymphocytes, using pan-centromeric and pan-telomeric probes. Int J Radiat Biol. 2004 Oct;80(10):737-44.
- Levy D, Vazquez M, Cornforth M, Loucas B, Sachs RK, Arsuaga J. Comparing DNA damage-processing pathways by computer analysis of chromosome painting data. J Comput Biol. 2004;11(4):626-41.
- Arsuaga J, Greulich-Bode KM, Vazquez M, Bruckner M, Hahnfeldt P, Brenner DJ, Sachs R, Hlatky L. Chromosome spatial clustering inferred from radiogenic aberrations. Int J Radiat Biol. 2004 Jul;80(7):507-15.
- Sachs RK, Levy D, Hahnfeldt P, Hlatky L. Quantitative analysis of radiation-induced chromosome aberrations. Cytogenet Genome Res. 2004;104(1-4):142-8. Review.
- Brenner DJ, Doll R, Goodhead DT, Hall EJ, Land CE, Little JB, Lubin JH, Preston DL, Preston RJ, Puskin JS, Ron E, Sachs RK, Samet JM, Setlow RB, Zaider M. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci USA. 2003 Nov 25;100(24):13761-6.
- Brenner DJ, Sachs RK. Domestic radon risks may be dominated by bystander effects--but the risks are unlikely to be greater than we thought. Health Phys. 2003 Jul;85(1):103-8.
- Hahnfeldt P, Folkman J, Hlatky L. Minimizing long-term tumor burden: the logic for metronomic chemotherapeutic dosing and its antiangiogenic basis. J Theor Biol. 2003 Feb 21;220(4):545-54.
- Vazquez M, Greulich-Bode KM, Arsuaga J, Cornforth MN, Brückner M, Sachs RK, Hlatky L, Molls M, Hahnfeldt P. Computer analysis of mFISH chromosome aberration data uncovers an excess of very complicated metaphases. Int J Radiat Biol. 2002 Dec;78(12):1103-15.
- Cornforth MN, Greulich-Bode KM, Loucas BD, Arsuaga J, Vázquez M, Sachs RK, Brückner M, Molls M, Hahnfeldt P, Hlatky L, Brenner DJ. Chromosomes are predominantly located randomly with respect to each other in interphase human cells. J Cell Biol. 2002 Oct 28;159(2):237-44.
- Sachs RK, Arsuaga J, Vázquez M, Hlatky L, Hahnfeldt P. Using graph theory to describe and model chromosome aberrations. Radiat Res. 2002 Nov;158(5):556-67.
- Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, Fletcher JA, Hlatky L, Hahnfeldt P, Folkman J, Kaipainen A. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002 Oct;110(7):923-32.
- Hlatky L, Sachs RK, Vazquez M, Cornforth MN. Radiation-induced chromosome aberrations: insights gained from biophysical modeling. Bioessays. 2002 Aug;24(8):714-23. Review.
- Brenner DJ, Sachs RK. Do low dose-rate bystander effects influence domestic radon risks? Int J Radiat Biol. 2002 Jul;78(7):593-604.
- Radivoyevitch T, Kozubek S, Sachs RK. The risk of chronic myeloid leukemia: can the dose-response curve be U-shaped? Radiat Res. 2002 Jan;157(1):106-9.
- Ponomarev AL, Sachs RK. Radiation breakage of DNA: a model based on random-walk chromatin structure. J Math Biol. 2001 Oct;43(4):356-76.
- Sachs RK, Hlatky LR, Hahnfeldt P. Simple ODE models of tumor growth and anti-angiogenic or radiation treatment, Math Comp Modelling 2001;33:1297-305.
- Radivoyevitch T, Sachs RK, Nikiforov YE, Nikiforova MN, Little MP. On target cell numbers in radiation-induced H4-RET mediated papillary thyroid cancer. Radiat Environ Biophys. 2001 Sep;40(3):191-7.
- Radivoyevitch T, Kozubek S, Sachs RK. Biologically based risk estimation for radiation-induced CML. Inferences from BCR and ABL geometric distributions. Radiat Environ Biophys. 2001 Mar;40(1):1-9.
- Ponomarev AL, Sachs RK. Radiation breakage of DNA: A model based on random-walk chromatin structure. J Math Biol. 2001;43:356-76.
- Ponomarev AL, Cucinotta FA, Sachs RK, Brenner DJ. Monte Carlo predictions of DNA fragment-size distributions for large sizes after HZE particle irradiation. Phys Med. 2001;17 Suppl 1:153-6.
- Ponomarev AL, Cucinotta FA, Sachs RK, Brenner DJ, Peterson LE. Extrapolation of the dna fragment-size distribution after high-dose irradiation to predict effects at low doses. Radiat Res. 2001 Nov;156(5 Pt 2):594-7.
- Costes S, Sachs R, Hlatky L, Vannais D, Waldren C, Fouladi B. Large-mutation spectra induced at hemizygous loci by low-LET radiation: evidence for intrachromosomal proximity effects. Radiat Res. 2001 Nov;156(5 Pt 1):545-57.
- Brenner DJ, Little JB, Sachs RK. The bystander effect in radiation oncogenesis: II. A quantitative model. Radiat Res. 2001 Mar;155(3):402-8.
- Sachs RK, Levy D, Chen AM, Simpson PJ, Cornforth MN, Ingerman EA, Hahnfeldt P, Hlatky LR. Random breakage and reunion chromosome aberration formation model; an interaction-distance version based on chromatin geometry. Int J Radiat Biol. 2000 Dec;76(12):1579-88.
- Brenner DJ, Sachs RK. Protraction effects in radiation studies: basic biophysics. Radiat Res. 2000 Dec;154(6):736-7.
- Ponomarev AL, Brenner D, Hlatky LR, Sachs RK. A polymer, random walk model for the size-distribution of large DNA fragments after high linear energy transfer radiation. Radiat Environ Biophys. 2000 Jun;39(2):111-20.
- Helmlinger G, Endo M, Ferrara N, Hlatky L, Jain RK. Formation of endothelial cell networks. Nature. 2000 May 11;405(6783):139-41.
- Sachs RK, Hlatky LR, Trask BJ. Radiation-produced chromosome aberrations: colourful clues. Trends Genet. 2000 Apr;16(4):143-6. Review.
- Sachs RK, Rogoff A, Chen AM, Simpson PJ, Savage JR, Hahnfeldt P, Hlatky LR. Underprediction of visibly complex chromosome aberrations by a recombinational-repair ('one-hit') model. Int J Radiat Biol. 2000 Feb;76(2):129-48.
- Ponomarev AL, Sachs RK. Polymer chromosome models and Monte Carlo simulations of radiation breaking DNA. Bioinformatics. 1999 Dec;15(12):957-64.
- Hahnfeldt P, Panigrahy D, Folkman J, Hlatky L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999 Oct 1;59(19):4770-5.
- Sachs RK, Ponomarev AL, Hahnfeldt P, Hlatky LR. Locations of radiation-produced DNA double strand breaks along chromosomes: a stochastic cluster process formalism. Math Biosci. 1999 Jul;159(2):165-87.
- Sachs RK, Chen AM, Simpson PJ, Hlatky LR, Hahnfeldt P, Savage JR. Clustering of radiation-produced breaks along chromosomes: modelling the effects on chromosome aberrations. Int J Radiat Biol. 1999 Jun;75(6):657-72.
- Brenner DJ, Sachs RK. A more robust biologically based ranking criterion for treatment plans. Int J Radiat Oncol Biol Phys. 1999 Feb 1;43(3):697-8.
- Hardenbergh PH, Hahnfeldt P, Hlatky L, Takemoto C, Shimamura A, McGill G, Fung CY, Bodis S, Fisher DE. Distinct mathematical behavior of apoptotic versus non-apoptotic tumor cell death. Int J Radiat Oncol Biol Phys. 1999 Feb 1;43(3):601-5.
- Hahnfeldt P, Hlatky L. Cell resensitization during protracted dosing of heterogeneous cell populations. Radiat Res. 1998 Dec;150(6):681-7.
- Sachs RK, Brenner DJ. The mechanistic basis of the linear-quadratic formalism. Med Phys. 1998 Oct;25(10):2071-3. Review.
- Sachs RK, Brenner DJ, Hahnfeldt PJ, Hlatky LR. A formalism for analyzing large-scale clustering of radiation-induced breaks along chromosomes. Int J Radiat Biol. 1998 Aug;74(2):185-206.
- Brenner DJ, Hlatky LR, Hahnfeldt PJ, Huang Y, Sachs RK. The linear-quadratic model and most other common radiobiological models result in similar predictions of time-dose relationships. Radiat Res. 1998 Jul;150(1):83-91.
- Radivoyevitch T, Hoel DG, Hahnfeldt P, Sachs RK. Size distributions of misrejoining DNA fragments in irradiated cells. Math Biosci. 1998 May;149(2):107-36. Review.
- Wu H, Sachs RK, Yang TC. Radiation-induced total-deletion mutations in the human hprt gene: a biophysical model based on random walk interphase chromatin geometry. Int J Radiat Biol. 1998 Feb;73(2):149-56.
- Radivoyevitch T, Hoel DG, Chen AM, Sachs RK. Misrejoining of double-strand breaks after X irradiation: relating moderate to very high doses by a Markov model. Radiat Res. 1998 Jan;149(1):59-67.
- Radivoyevitch T, Hoel DG, Hahnfeldt PJ, Rydberg B, Sachs RK. Recent data obtained by pulsed-field gel electrophoresis suggest two types of double-strand breaks. Radiat Res. 1998 Jan;149(1):52-8.
- Chen AM, Lucas JN, Simpson PJ, Griffin CS, Savage JR, Brenner DJ, Hlatky LR, Sachs RK. Computer simulation of data on chromosome aberrations produced by X rays or alpha particles and detected by fluorescence in situ hybridization. Radiat Res. 1997 Nov;148(5 Suppl):S93-101.
- Sachs RK, Hahnfeldt P, Brenner DJ. The link between low-LET dose-response relations and the underlying kinetics of damage production/repair/misrepair. Int J Radiat Biol. 1997 Oct;72(4):351-74. Review.
- Sachs RK, Brenner DJ, Chen AM, Hahnfeldt P, Hlatky LR. Intra-arm and interarm chromosome intrachanges: tools for probing the geometry and dynamics of chromatin. Radiat Res. 1997 Oct;148(4):330-40. Review.
- Wu H, Durante M, Sachs RK, Yang TC. Centric rings, acentric rings and excess acentric fragments based on a random-walk interphase chromosome model. Int J Radiat Biol. 1997 May;71(5):487-96.
- Liu B, Sachs RK. A two-backbone polymer model for interphase chromosome geometry. Bull Math Biol. 1997 Mar;59(2):325-37.
- Sachs RK, Chen AM, Brenner DJ. Review: proximity effects in the production of chromosome aberrations by ionizing radiation. Int J Radiat Biol. 1997 Jan;71(1):1-19. Review.
- Sachs RK, Heidenreich WF, Brenner DJ. Dose timing in tumor radiotherapy: considerations of cell number stochasticity. Math Biosci. 1996 Dec;138(2):131-46. Review.
- Brenner DJ, Hahnfeldt P, Amundson SA, Sachs RK. Interpretation of inverse dose-rate effects for mutagenesis by sparsely ionizing radiation. Int J Radiat Biol. 1996 Oct;70(4):447-58.
- Brenner DJ, Sachs RK. Comments on "Comment on the ratio of chromosome-type dicentric interchanges to centric rings for track-clustered compared with random breaks" by Savage and Papworth (Radiat. Res. 146, 236-240, 1996). Radiat Res. 1996 Aug;146(2):241-2.
- Hlatky L, Hahnfeldt P, Tsionou C, Coleman CN. Vascular endothelial growth factor: environmental controls and effects in angiogenesis. Br J Cancer Suppl. 1996 Jul;27:S151-6. Review.
- Chen AM, Lucas JN, Hill FS, Brenner DJ, Sachs RK. Proximity effects for chromosome aberrations measured by FISH. Int J Radiat Biol. 1996 Apr;69(4):411-20.
- Hlatky L, Olesiak M, Hahnfeldt P. Measurement of potential doubling time for human tumor xenografts using the cytokinesis-block method. Cancer Res. 1996 Apr 1;56(7):1660-3.
- Lucas JN, Chen AM, Sachs RK. Theoretical predictions on the equality of radiation-produced dicentrics and translocations detected by chromosome painting. Int J Radiat Biol. 1996 Feb;69(2):145-53.
- Hahnfeldt P, Hlatky L. Resensitization due to redistribution of cells in the phases of the cell cycle during arbitrary radiation protocols. Radiat Res. 1996 Feb;145(2):134-43.
- Chen AM, Lucas JN, Hill FS, Brenner DJ, Sachs RK. Chromosome aberrations produced by ionizing radiation: Monte Carlo simulations and chromosome painting data. Comput Appl Biosci. 1995 Aug;11(4):389-97.
- Brenner DJ, Hlatky LR, Hahnfeldt PJ, Hall EJ, Sachs RK. A convenient extension of the linear-quadratic model to include redistribution and reoxygenation. Int J Radiat Oncol Biol Phys. 1995 May 15;32(2):379-90.
- Hlatky L, Van Buren T, Hahnfeldt P. Quantifying intracellular radioresponse diversity in irradiated sandwich cultures via micronucleus expression. Int J Radiat Biol. 1995 May;67(5):541-8.
- Chen PL, Brenner DJ, Sachs RK. Ionizing radiation damage to cells: effects of cell cycle redistribution. Math Biosci. 1995 Apr;126(2):147-70.
- Sachs RK, van den Engh G, Trask B, Yokota H, Hearst JE. A random-walk/giant-loop model for interphase chromosomes. Proc Natl Acad Sci USA. 1995 Mar 28;92(7):2710-4. PMC42288.
- Hahnfeldt P, Hlatky LR, Brenner DJ, Sachs RK. Chromosome aberrations produced by radiation: the relationship between excess acentric fragments and dicentrics. Radiat Res. 1995 Feb;141(2):136-52.
- Brenner DJ, Hall EJ, Huang Y, Sachs RK. Potential reduced late effects for pulsed brachytherapy compared with conventional LDR. Int J Radiat Oncol Biol Phys. 1995 Jan 1;31(1):201-2.
- Brenner DJ, Sachs RK. Chromosomal "fingerprints" of prior exposure to densely ionizing radiation. Radiat Res. 1994 Oct;140(1):134-42.
- Hlatky LR, Hahnfeldt P, Sachs RK. Influence of time-dependent stochastic heterogeneity on the radiation response of a cell population. Math Biosci. 1994 Aug;122(2):201-20.
- Brenner DJ, Hall EJ, Huang Y, Sachs RK. Optimizing the time course of brachytherapy and other accelerated radiotherapeutic protocols. Int J Radiat Oncol Biol Phys. 1994 Jul 1;29(4):893-901. Review.
- Hahnfeldt P, Hlatky LR. A Monte Carlo/Markov chain model for the association of data for chromosome aberrations and formation of micronuclei. Radiat Res. 1994 May;138(2):239-45.
- Chen PL, Sachs R. Singular perturbation theory applied to Markov models for DNA damage caused by ionizing radiation. J Theor Biol. 1994 Jan 21;166(2):117-26.
- Brenner DJ, Ward JF, Sachs RK. Track structure, chromosome geometry and chromosome aberrations. Basic Life Sci. 1994;63:93-109; discussion 109-13.
- Sachs RK, Brenner DJ. Effect of LET on chromosomal aberration yields. I. Do long-lived, exchange-prone double strand breaks play a role? Int J Radiat Biol. 1993 Dec;64(6):677-88.
- Hahnfeldt P, Hearst JE, Brenner DJ, Sachs RK, Hlatky LR. Polymer models for interphase chromosomes. Proc Natl Acad Sci USA. 1993 Aug 15;90(16):7854-8.
- Hurwitz SJ, Hlatky L. Assessment of radiation response on a cell-by-cell basis using in situ densitometric imaging of micronuclei. Radiat Res. 1993 Apr;134(1):112-6.
- Sachs RK, Awa A, Kodama Y, Nakano M, Ohtaki K, Lucas JN. Ratios of radiation-produced chromosome aberrations as indicators of large-scale DNA geometry during interphase. Radiat Res. 1993 Mar;133(3):345-50.
- Lucas JN, Sachs RK. Using three-color chromosome painting to test chromosome aberration models. Proc Natl Acad Sci USA. 1993 Feb 15;90(4):1484-7.
- Sachs RK, Chen P, Hahnfeldt P, Lai D, Hlatky LR. DNA damage in non-proliferating cells subjected to ionizing irradiation at high or low dose rates. J Math Biol. 1993;31(3):291-315.
- Trask BJ, Allen S, Massa H, Fertitta A, Sachs R, van den Engh G, Wu M. Studies of metaphase and interphase chromosomes using fluorescence in situ hybridization. Cold Spring Harb Symp Quant Biol. 1993;58:767-75.
- Sachs RK, Chen PL, Hahnfeldt PJ, Hlatky LR. DNA damage caused by ionizing radiation. Math Biosci. 1992 Dec;112(2):271-303.
- Sachs RK, Yates BL, Tarver J, Morgan WF. Modelling the formation of polycentric chromosome aberrations. Int J Radiat Biol. 1992 Oct;62(4):449-60.
- van den Engh G, Sachs R, Trask BJ. Estimating genomic distance from DNA sequence location in cell nuclei by a random walk model. Science. 1992 Sep 4;257(5075):1410-2.
- Hlatky LR, Sachs RK, Hahnfeldt P. The ratio of dicentrics to centric rings produced in human lymphocytes by acute low-LET radiation. Radiat Res. 1992 Mar;129(3):304-8.
- Hahnfeldt P, Sachs RK, Hlatky LR. Evolution of DNA damage in irradiated cells. J Math Biol. 1992;30(5):493-511.
- Hlatky L, Sachs R, Hahnfeldt P. Reaction kinetics for the development of radiation-induced chromosome aberrations. Int J Radiat Biol. 1991 May;59(5):1147-72.
- Sachs RK, Hlatky L, Hahnfeldt P, Chen PL. Incorporating dose-rate effects in Markov radiation cell survival models. Radiat Res. 1990 Nov;124(2):216-26.
- Hlatky L, Ring CS, Sachs RK. Detection of an intrinsic marker in hypoxic cells. Cancer Res. 1989 Sep 15;49(18):5162-6.
- Hlatky L, Ring C, Sachs RK. Comparison of 3H-misonidazole binding between CHO and 9L cells using the sandwich system. Int J Radiat Oncol Biol Phys. 1989 Apr;16(4):943-7.
- Hlatky L, Sachs RK, Ring CS. Reducing the hypoxic fraction of a tumour model by growth in low glucose. Br J Cancer. 1989 Mar;59(3):375-80.
- Hlatky L, Ring CS, Sachs RK. 3H-misonidazole labeling and viability of hypoxic cells in the sandwich system, an in vitro tumor analogue. Int J Radiat Oncol Biol Phys. 1989 Jan;16(1):143-53.
- Hlatky L, Sachs RK, Alpen EL. Joint oxygen-glucose deprivation as the cause of necrosis in a tumor analog. J Cell Physiol. 1988 Feb;134(2):167-78.
- Hlatky L, Alpen EL, Yee MK. Differences in the X-ray sensitivity of cells in different regions of the sandwich, a diffusion-limited system for cell growth. Radiat Res. 1986 Oct;108(1):62-73.
- Hlatky L, Alpen EL. Two-dimensional diffusion limited system for cell growth. Cell Tissue Kinet. 1985 Nov;18(6):597-611.